
Iñaki Comas (Valencia, 1979) es biólogo y experto en genómica. En el laboratorio del Instituto de Biomedicina de Valencia (IBV-CSIC) donde trabaja, combina la información genómica de patógenos con datos epidemiológicos para estudiar el desarrollo y evolución de las enfermedades infecciosas en las poblaciones.
Hasta hace unos meses, el trabajo de Iñaki estaba enfocado en estudiar formas de controlar la propagación de la tuberculosis y determinar qué factores genéticos influyen en la virulencia de su agente causal, la bacteria Mycobacterium tuberculosis. No obstante, desde la llegada del brote de COVID-19, causado por el coronavirus SARS-CoV-2, sus investigaciones se han trasladado al estudio de este virus.
En la actualidad, Iñaki Comas lidera, junto a Fernando González, investigador del Instituto de Biología Integrativa de Sistemas (I2SysBio) del CSIC y la Universitat de València, y la colaboración de la Fundación para el Fomento de la Investigación Sanitaria y Biomédica (FISABIO) de la Generalitat Valenciana, un proyecto del CSIC destinado a estudiar la evolución de la epidemia de COVID.
Desde Genotipia, hemos hablado con Iñaki Comas para aprender sobre la epidemiología genómica y conocer cómo puede ser utilizada para controlar la actual epidemia de COVID-19 y otras futuras epidemias.

¿Qué es la epidemiología genómica?
Es un poco la evolución de la epidemiología molecular, que consiste en usar marcadores genéticos para identificar parecidos entre muestras de un mismo patógeno. En este caso, la innovación es que ahora tenemos acceso a hacerlo a gran escala y con genomas completos. Antes, cuando usábamos solo unos fragmentos del genoma para hacer la epidemiología molecular, se accedía a una parte muy pequeña del genoma. Ahora podemos acceder a genomas completos, lo que nos da una resolución mucho mayor.
El acceso al genoma completo como marcador, los métodos que permiten combinar información del genoma y diversidad de las muestras con datos epidemiológicos y el hecho de que poco a poco estamos llegando a secuenciar casi en tiempo real, están llevando a que la epidemiología genómica sea un campo que permite hacer una vigilancia epidemiológica prácticamente en tiempo real. Detecta con mucha precisión la aparición de brotes de diferentes enfermedades infecciosas. Actualmente, es el caso del coronavirus, pero algo parecido es lo que hacemos en tuberculosis o, la resistencia a Klebsiella en hospitales que estudia Fernando González. Lo importante es que ahora tenemos una precisión mucho más grande y poco a poco se empieza a hacer en tiempo real, lo que permite tener una respuesta más rápida.
¿Y el estudio del genoma de los hospedadores?
Técnicamente también está incluido en la epidemiología genómica, aunque esta se ha centrado sobre todo en los patógenos, que es donde más se aplica. De hecho, hemos planteado en tuberculosis y también en coronavirus, que lo que necesitaríamos sería secuenciar patógeno y hospedador. De esta manera siempre se sabría cuál es el genoma del hospedador asociado al genoma del patógeno. Esto permitiría, por una parte, hacer todos los análisis de epidemiologia genómica con los patógenos, y después, con la información del hospedador, hacer otros estudios. Además, hoy en día hay técnicas que permiten hacer asociaciones genéticas usando el genoma del hospedador y el genoma del virus.
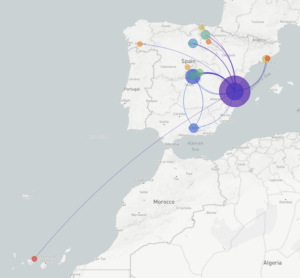
Lideras junto a Fernando González un proyecto que estudiará los genomas del coronavirus SARS-CoV-2 tomados de muestras de pacientes infectados. ¿Podrías indicarnos en qué consiste este proyecto y cuál es su objetivo?
El proyecto intenta estudiar los genomas del coronavirus de una muestra representativa de pacientes de toda España. Para ello lo que hemos hecho es montar un consorcio en el que están implicados los grandes hospitales del país. En el consorcio, el papel de las Unidades de Microbiología Clínica está siendo fundamental para tener una representatividad de lo que está ocurriendo en toda España. Aunque ahora mismo están desbordadas, están haciendo un esfuerzo muy grande para proveer de muestras o capacidad de secuenciación.
Una vez tenemos las muestras, secuenciamos el genoma del coronavirus de los pacientes y miramos las diferencias genéticas entre los virus que infectan a diferentes pacientes. Con esta información inferimos los patrones de transmisión, cómo se ha ido transmitiendo el virus en el tiempo y a nivel geográfico.
Los datos que obtengáis serán depositados en bases de datos públicas y en la plataforma NextStrain. De hecho, habéis creado una página derivada de Nextstrain que permite ver la evolución del coronavirus en España, en el espacio y en el tiempo (COVID-19 Spain). Ahora mismo hay 85 genomas estudiados ¿Cuántos aspiráis a conseguir? ¿Cuál sería una muestra representativa?
En la primera parte del proyecto, para la que tenemos financiación de momento, la idea es ver qué ocurrió en España en las primeras semanas de la epidemia. Para ello estamos intentando secuenciar alrededor de tres mil o cuatro mil casos. Queremos saber también por qué en algunas zonas el virus se expandió mucho más que en otras zonas. Por esto, en algunas zonas especialmente interesantes, como por ejemplo La Rioja o Álava, que no tienen un número absoluto de casos muy elevado pero sí un tienen un numero relativo muy grande, incrementaremos el esfuerzo de muestreo.
Sabemos que al principio veremos muchas importaciones desde China, Italia y otros sitios que tenían el virus. Poco a poco deberíamos ver viendo cómo se va expandiendo en España y cómo ocurrió la transmisión comunitaria. Cada región de España ha tenido epidemiología diferente y queremos entender por qué.
¿Cuánto tiempo se tarda en secuenciar uno de estos genomas?
Con personal dedicado y disponibilidad de máquinas y reactivos se tarda entre tres y cuatro días con tecnología Nanopore y siete días con Illumina. Esta última permite incluir muchas más muestras. Dependiendo de la necesidad elegimos una plataforma u otra.
Con la información de los datos de secuenciación del coronavirus en España y otros países ¿cómo ha cambiado el genoma del virus desde el inicio del brote?
Una noticia relativamente buena es que no tiene una tasa de mutación muy alta, para ser un virus de ARN. Sabemos que hay otros virus de ARN que tienen una tasa bastante más alta. Aunque es verdad que estamos al principio de la epidemia y calcular esas tasas es complicado porque se está expandiendo ahora mismo y no sabemos si la tasa está estabilizada.
Se producen cambios pero no hemos visto que se produzcan en zonas que parezcan relevantes para la infección o que puedan ser de escape al sistema inmunitario. Sí pensamos que acabará pasando. Por ejemplo, cuando entren nuevos antivirales o vacunas, el virus igual consigue escapar de los primeros con mutaciones de resistencia o de las vacunas con mutaciones como las que ocurren en el virus de la gripe. No estamos seguros si por eso es importante mantener el esfuerzo en el tiempo para monitorizar todo este tipo de eventos.
¿Es posible que el virus se haga menos virulento?
Ahora mismo hay diferentes teorías sobre este tema. Algunos piensan que terminará haciéndose menos virulento. Aunque generalmente, los virus que se hacen menos virulentos suelen aumentar su transmisión y no creo que este pueda aumentar su tasa de transmisión. Ya se transmite bastante bien. Todavía no está claro. Dependerá mucho de la inmunidad de grupo que se genere, que tampoco sabemos si se generará o cuánto tiempo durará. También dependerá de la existencia de una vacuna.
Haz mencionado que uno de los retos del estudio es obtener resultados que sirvan a las autoridades de Salud Pública. ¿Pueden estos datos ser utilizados para plantear medidas de control de la propagación de la actual pandemia? ¿Cómo?
Por una parte, si mantenemos la secuenciación sostenida en el tiempo podemos informar a la Salud Pública sobre las medidas de contención. Por ejemplo, si podemos ver como se está moviendo el virus por España y se imponen medidas de restricción como las que tenemos ahora, deberíamos ver que el virus cada vez se transmite menos entre regiones de España. Si no es así, es que están fallando en algún sitio las medidas de contención.
Otra información importante, aunque sea en retrospectiva, es entender en qué momento, al principio de la epidemia, el virus pasó de ser importado a transmitirse comunitariamente. En el futuro nos va a volver a pasar. El virus va a volver a introducirse en el país. Más la presencia del virus que permanezca aquí, que no sabemos cómo será. Es importante distinguir cuándo tenemos casos importados y cuándo tenemos casos que se transmiten comunitariamente y son difíciles de identificar. Es lo que ha pasado al inicio de la epidemia. Se pensó que la mayor parte de los casos eran importados y estaban controlados y se vio que, por ejemplo en Madrid, había muchos casos de transmisión comunitaria que no habían sido detectados. Nosotros podemos analizarlo e intentar identificar indicadores que nos digan que eso está pasando.
Los datos obtenidos, ¿serán también de utilidad para otras epidemias?
La ambición, no solo del proyecto, sino del país, debería ser establecer plataformas de respuesta rápida a pandemias. No solo de secuenciación de patógenos u otros aspectos epidemiológicos, sino en otros ámbitos, como económico o sanitario. Ahora mismo se están tomando medidas a toda prisa porque no existían antes.
A nivel científico hay que tener en cuenta que si se quiere una respuesta rápida a una pandemia debe haber equipos con cierta estabilidad. No puede ser que vivamos siempre en la temporalidad. En mi equipo, el único indefinido soy yo. En el de Fernando creo que solo él. Si se pide a la ciencia que dé respuestas rápidas, no puede ser que dependa de equipos con contratos de meses.
Tu equipo trabaja también con Mycobacterium tuberculosis, la bacteria responsable de causar tuberculosis. ¿Qué retos ha presentado estudiar un virus como SARS-CoV2, frente a una bacteria?
Es curioso, porque tienen bastantes cosas comunes. Evidentemente una bacteria es mucho más grande que un virus y por lo tanto permite muestras más grandes. Por otra parte, la tasa de evolución de Mycobacterium tuberculosis es mucho más lenta, pero tiene un genoma más grande, por lo que al final el número de mutaciones no es tan diferente. También es muy interesante que las dos sean afecciones respiratorias y las dos parezca que se transmiten durante estadios asintomáticos. Además, las medidas de control son muy parecidas. El seguimiento de contactos, identificar casos activos…son las medidas que se usan en la actualidad en Valencia para la tuberculosis.
Luego cada uno tiene sus características. Por ejemplo, este virus afecta sobre todo a vías respiratorias altas y luego puede pasar a vías bajas, mientras que tuberculosis afecta a las vías bajas. Además, a la hora de modelar la epidemiología de la enfermedad, la infección por tuberculosis tiene un estadio de latencia mucho más grande. Mucha gente está infectada y no lo sabe porque el sistema inmunitario lo controla. En virus ese estadio es mucho más agudo y todo ocurre en días o como mucho meses.
Precisamente este año finaliza un proyecto financiado por la Unión Europea del cual eres Investigador Principal, destinado a acelerar la erradicación de la tuberculosis. ¿Puedes avanzarnos algunos de sus resultados?
Hace nada hemos publicado algo muy relacionado con el coronavirus, que es intentar entender si se puede transmitir la enfermedad en estadios asintomáticos. Gracias a las técnicas de epidemiologia genómica que he comentado hemos demostrado que eso pasa en tuberculosis. Hay gente que tiene tuberculosis, no tiene prácticamente síntomas y es capaz transmitirla. Eso tiene una serie de implicaciones en el control de la tuberculosis porque se asume siempre que solo una persona con síntomas muy claros puede transmitir la enfermedad. Nosotros hemos visto que no es así. Otros investigadores con otras aproximaciones como imagen médica y análisis de expresión también han demostrado que hay personas que parecen sanas pero pueden ser consideradas casos activos.
Desde el punto de vista de control, esto implica que en lugar de esperar que aparezcan los casos en la clínica hay que buscarlos activamente. De hecho, aunque hay pocos casos en España, lo que vemos es que muchos de ellos son de transmisión. No es gente que se infectó hace 20 años en los que se ha reactivado la enfermedad.
En países de alta incidencia, la transmisión es todavía más importante. Entender que hay un estadio presintomático en el que puedes transmitir la enfermedad es importante porque cambia los modelos de cómo deberíamos de controlar la tuberculosis.
Tuberculosis, COVID-19… ¿Es la epidemiología genómica la herramienta de control para las epidemias futuras?
Yo tengo esta frase de que la epidemiología genómica es para el siglo XXI lo que fueron las vacunas para el siglo XIX y los antibióticos al siglo XX. Lo que quiero decir con ella es que la epidemiología genómica en tiempo real es un salto de innovación como lo fueron en su momento las vacunas y los antibióticos. La epidemiología genómica va a jugar un papel importante para detectar la emergencia de nuevos virus e identificar la transmisión en estadios muy tempranos de enfermedades infecciosas, como COVID-19. Sobre todo cuando consigamos acoplarlo todo a una secuenciación en tiempo real y a un traslado de esos resultados, prácticamente en tiempo real, a las autoridades que tienen que tomar decisiones. Está pasando y creo que a lo largo de los próximos años lo vamos a ver más. Hay países que tienen esto muy presente, como Reino Unido.
Precisamente el Instituto Wellcome Sanger anunció que iba a secuenciar de forma masiva el genoma del coronavirus de los pacientes de Reino Unido.
El Reino Unido lleva apostando unos años por la secuenciación genómica en el sistema de salud, no solo en cuanto a patógenos sino también en pacientes. Esto permite saber muchas más cosas de las que vemos nosotros ahora. Nosotros estamos viendo las cosas que veíamos en el siglo XX mientras que ellos ya están viendo cosas del siglo XXI. Poco a poco vamos en esa dirección y hay grupos en tanto en genética humana como en patógenos que siguen ese camino. Lo que falta es una apuesta más fuerte del Estado para construir estructuras o plataformas más estables.
Amparo Tolosa, Genotipia
Fuente: https://genotipia.com/genetica_medica_news/inaki-comas-entrevista-epidemiologia-genomica/




