
17 de mayo: el Día de las Letras Gallegas, el Día de la Constitución Noruega y, finalmente, también el Día Mundial de Concienciación sobre la Neurofibromatosis.
Descrita por primera vez en 1882 por Friedrich Daniel von Recklinghausen, esta enfermedad (o, más bien, grupo de enfermedades) aparece en uno de cada 2.000 nacimientos. No suele ser letal por sí sola, pero sí que reduce la esperanza de vida y sus efectos pueden ser negativos para el bienestar de los pacientes.
La neurofibromatosis se caracteriza por la aparición de tumores benignos (bultos) a lo largo de nervios y debajo de la piel. Los tumores suelen causar problemas neurológicos como dolor, sordera o ceguera. Y si se encuentran debajo de la piel, también pueden aparecer bultos sobre la superficie del cuerpo.
¿Qué es la neurofibromatosis?
La neurofibromatosis es una enfermedad genética, autosómica (se encuentra en los cromosomas que no están relacionados con el sexo) y dominante (heredar una sola copia mutada del gen es suficiente para que se manifieste la enfermedad). Según el tipo de neurofibromatosis la mutación que causa la enfermedad se encuentra en un gen diferente.
En la mitad de los casos el paciente ha heredado la mutación de uno de sus padres, mientras que en la otra es de novo, no presentándose en los padres. En cualquier caso, una persona con fibromatosis ya nace con ella, no se puede contagiar. Además, la neurofibromatosis no es una enfermedad tiquismiquis: no distingue entre razas ni géneros.
Tipos de Neurofibromatosis
En realidad, el término neurofibromatosis incluye 3 enfermedades:
- Neurofibromatosis tipo 1 (Nf1): también llamada “enfermedad de Von Recklinghausen”. En este caso el gen alterado es NF1, que se encuentra en el cromosoma 17. Este tipo de neurofibromatosis se caracteriza por la ocurrencia de cambios en la piel (manchas de color café con leche y pecas bajo la axila) y bultos del tamaño de un guisante por la superficie del cuerpo, llamados neurofibromas. En algunos casos también se forman unas masas de pigmento sobre el iris del ojo (nódulos de Lisch) o incluso deformación del esqueleto. Por último, aunque la inteligencia de los niños con Nf1 es normal, en algunos casos pueden tener problemas de memoria, atención, orientación espacial y procesado de información.
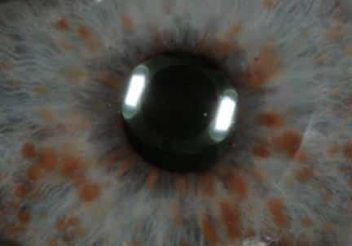
Nódulos de Lisch sobre el iris, típicos de la Nf1
- Neurofibromatosis tipo 2 (Nf2): también llamada neurofibromatosis acústica lateral. En este caso el gen alterado (NF2) se encuentra en el cromosoma 22. Se caracteriza por pérdidas de audición, cataratas y problemas de equilibrio, además de los bultos mencionados en la Nf1
- Schwannomatosis: esta es la variante más rara de neurofibromatosis, y sólo se ha descubierto recientemente. Está causada por la mutación de un gen llamado SMARCB1 o uno llamado INI1, localizados también en el cromosoma 22. El síntoma principal es el dolor en todas partes del cuerpo, pero también pueden ocurrir cosquilleos, debilidad en los dedos e insensibilidad al tacto.
Hay que decir que todas estas enfermedades presentan mucha variabilidad: no hay dos pacientes iguales, e incluso se pueden ver enormes diferencias entre familiares.
Curiosamente, a diferencia de la mayoría de enfermedades dominantes, en la neurofibromatosis se necesitan dos copias del gen alterado, en lugar de una, para que se manifieste la enfermedad. Sin embargo se considera dominante. ¿Cómo puede ser? Resulta que cuando una persona ya tiene una copia mutada (ya sea heredada o de novo), en la mayoría de los casos se produce una segunda mutación en la otra copia del gen en cada una de las células especializadas que rodean los nervios. Se necesita esta segunda mutación para que se puedan formar los tumores. De esta forma, la neurofibromatosis es una enfermedad que al mismo tiempo necesita una y dos mutaciones. Una enfermedad dominante en herencia y recesiva en el desarrollo de los síntomas.
El origen neuronal
Los genes responsables de la neurofibromatosis contienen la información para que las células fabriquen unas proteínas supresoras de tumores. En una situación normal estas proteínas regulan la división y el crecimiento celular, impidiendo que se formen tumores. En pacientes con neurofibromatosis, como los genes responsables han sido alterados, estas proteínas no se pueden producir correctamente y las células seguirán multiplicándose y creciendo hasta formar una masa llamada tumor.
Como estos genes se expresan en neuronas y otras células nerviosas, los pacientes con neurofibromatosis desarrollarán tumores en estructuras y órganos donde se encuentran estas células, como el cerebro, la médula espinal, o en cualquier nervio.
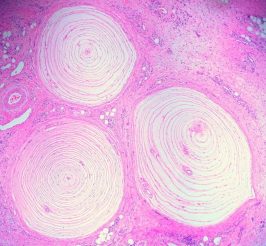
Corte transversal de fibras nerviosas, mucho más gruesas de lo normal.
Todos los síntomas se explican con la aparición de tumores: en la neurofibromatosis tipo 1 los bultos en la piel son tumores que se han formado en los nervios del tacto y la deformación ósea ocurre por la presión de tumores adyacentes. En la neurofibromatosis tipo 2 las pérdidas en los sentidos se explican por la formación de schwannomas (hinchazón de las células de Schwann, que rodean las neuronas con una vaina protectora de proteína para acelerar la señal nerviosa) alrededor del nervio que comunica con los ojos y oídos. En la schwannomatosis se forman schwannomas alrededor de los otros nervios, haciendo que duela.
Los tumores son benignos, por lo que no hay riesgo de que los tumores se trasladen a otros tejidos.
Diagnóstico y tratamiento
Debido a que existe tanta variabilidad entre los pacientes, puede resultar algo complicado diagnosticar la enfermedad. El diagnóstico se hace mediante examinación física del paciente, buscando un número mínimo de síntomas característicos mediante un criterio estándar. Los síntomas suelen aparecer durante la infancia en el caso de la neurofibromatosis tipo 1, durante la adolescencia en el caso de la tipo 2 y entre los 30 y los 60 años en el caso de la schwannomatosis. Si aún existen dudas sobre la enfermedad, siempre se puede hacer un análisis genético.
Por desgracia, no existe cura para la neurofibromatosis. El tratamiento consiste en calmar los síntomas: cirugía o radiación para eliminar los tumores, audífonos para la pérdida de audición, etc.

Vivir con la enfermedad
La neurofibromatosis no es una discapacidad, pues no impide vivir con normalidad: el paciente puede estudiar, trabajar y salir perfectamente sin ayuda. Sin embargo, sí que pueden verse afectados en el terreno social, por ejemplo, debido a las deformaciones físicas y a los bultos que pueden crecer sobre la superficie del cuerpo.
También hay que tener en cuenta que esta enfermedad puede ser un factor de riesgo que haga más fácil la aparición de otras. Aunque los tumores de la neurofibromatosis son benignos, sí que existe una mayor proporción de pacientes que desarrollan cáncer, en comparación con el resto de la población. De hecho, como hemos dicho al principio, la esperanza de vida en pacientes con neurofibromatosis suele ser menor.
Se puede realizar un estudio familiar para determinar si la mutación es heredada o de novo. Como la enfermedad se presenta en el 100% de los adultos que presenten la mutación, unos padres asintomáticos que hayan tenido un hijo con neurofibromatosis tienen las mismas probabilidades que el resto de la población normal de tener otro con la enfermedad. En cambio, cada uno de los hijos de un paciente con NF tiene un 50% de posibilidades de sufrirla también.

Artículos relacionados: neurofibromatosis, tumores benignos, enfermedad genética
Fuente: https://goo.gl/XFMDjV




