
España:
El síndrome de Sjögren-Larsson es una rara enfermedad genética con una prevalencia estimada de 1/250.000, caracterizada por ictiosis acompañada de prurito, trastornos neurológicos y afectación oftalmológica. Desde el punto de vista neurológico, este síndrome se engloba dentro de las leucodistrofias, alteraciones usualmente hereditarias de la mielina que afectan de forma predominante a la sustancia blanca del sistema nervioso central. Sjögren y Larsson describieron por primera vez este trastorno en 1957 en una serie de 28 pacientes con ictiosis, trastornos espásticos y oligofrenia [1].
En el nacimiento, que suele ser prematuro, se observa eritrodermia e hiperqueratosis leve, que evoluciona a una ictiosis generalizada que afecta con más gravedad a las zonas de flexión, la nuca, el tronco y las extremidades. A diferencia de otras formas de ictiosis, la afectación dermatológica se acompaña de un prurito de moderado a intenso. Las manifestaciones neurológicas se desarrollan durante los primeros años de vida. La mayoría de los pacientes sufre paresia espástica bilateral, que afecta más a los miembros inferiores. Son frecuentes la disartria y el retraso en el habla. En un 40% de los casos se producen convulsiones. Los pacientes presentan una leucodistrofia más o menos grave en la neuroimagen. La resonancia magnética muestra afectación difusa de la sustancia blanca, especialmente periventricular, frontal, parietal, de cuerpo calloso, y corona radiada con fibras en U preservadas. El retraso mental es de moderado a grave. Es frecuente la afectación oftalmológica, que puede incluir fotofobia, disminución de la agudeza visual, miopía e inclusiones cristalinas en la retina alrededor de la fóvea. Estas inclusiones son casi patognomónicas del síndrome de Sjögren-Larsson, pero no se observan en todos los pacientes [2–4].
El síndrome de Sjögren-Larsson es una enfermedad genética (OMIM 270200) de herencia autosómica recesiva causada por mutaciones en el gen ALDH3A2 (17p11.2), que codifica la enzima aldehído graso deshidrogenasa (FALDH). Estas mutaciones originan la pérdida de actividad de la enzima FALDH, implicada en el metabolismo lipídico y presente en todos los tipos celulares [2,3]. Los síntomas de la enfermedad surgen por la acumulación de alcoholes grasos y sus productos lipídicos metabólicos, lo cual causan una pérdida no específica de mielina en el sistema nervioso, toxicidad celular y disfunción del estrato córneo de la piel. El metabolismo del leucotrieno B4, mediador proinflamatorio, está también afectado, lo que explicaría el prurito grave que sufren estos pacientes y diferencia el síndrome de Sjögren-Larsson de otras ictiosis [5].
El diagnóstico del síndrome de Sjögren-Larsson originalmente se realizaba mediante la determinación de la actividad de la FALDH en fibroblastos cultivados a partir de una biopsia de piel o por los niveles elevados de leucotrieno B4 en la orina. Debido a la dificultad para acceder a estas pruebas, en la actualidad se recomienda el análisis de la secuencia del gen ALDH3A2 en pacientes con sospecha clínica en búsqueda de mutaciones [3].
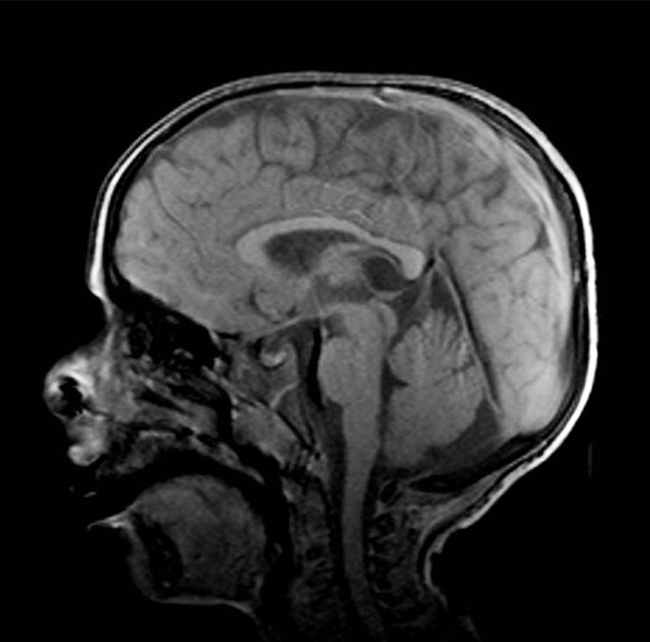
Varón de 5 años nacido mediante cesárea a las 36 semanas de gestación con peso de 2.900 g, sin antecedentes familiares de enfermedad cutánea o neurológica conocidos y con consanguinidad lejana entre ambos progenitores. A los 7 meses presentó piel seca y escamosa, y se le diagnosticó ictiosis. El resultado de la anatomía patológica reveló hiperqueratosis, presencia de capa granular y leve acantosis e infiltrado linfocitario perivascular superficial. A los 22 meses fue evaluado por neuropediatría al presentar retraso en el desarrollo psicomotor, y se observó espasticidad de los miembros inferiores con alteraciones de la marcha. La resonancia magnética cerebral mostró una afectación bilateral y asimétrica de la sustancia blanca que afectaba tanto a la sustancia blanca profunda próxima a los atrios ventriculares como a la sustancia blanca subcortical con preservación de las fibras en U (Figura). En la exploración oftalmológica presentó estrabismo, miopía y astigmatismo, con una agudeza visual corregida de 0,7 en el ojo derecho y de 0,3 en el izquierdo. No se observó fotofobia. La oftalmoscopia fue normal, sin distrofia macular. Ante la sospecha de síndrome de Sjögren-Larsson, se remitió al paciente a la unidad de genética para un estudio genético-molecular de ALDH3A2 mediante secuenciación. Los resultados revelaron la presencia de la variante c.991G>A (p.Glu331Lys) en homocigosis, lo que confirmó el diagnóstico clínico de síndrome de Sjögren-Larsson.
En el caso descrito, los hallazgos son similares a los casos publicados en la bibliografía: nacimiento prematuro, ictiosis congénita, prurito, retraso madurativo, espasticidad de los miembros inferiores y alteraciones en la sustancia blanca del sistema nervioso. La ictiosis, el prurito y la espasticidad están presentes en el 100% de los pacientes, y la afectación oftalmológica es variable [2]. En cuanto a la afectación oftalmológica, sólo se observó miopía y estrabismo, y no se detectó fotofobia ni maculopatía. Según algunos autores, esta maculopatía se observa sólo en un tercio de los pacientes; sin embargo, estudios que utilizan técnicas de alta resolución para el estudio de la retina, como la angiografía o la tomografía de coherencia óptica, detectan depósitos hiperrefringentes en la totalidad de los casos de síndrome de Sjögren-Larsson estudiados [4,6]. Es probable que la maculopatía cristalina esté presente en el 100% de los pacientes con distintos grados de gravedad. En los casos en los que no se aprecia, como el aquí presentado, podría tratarse de una forma leve no detectable mediante un examen convencional de fondo de ojo. La edad del paciente es otro factor a tener en cuenta, ya que el número de inclusiones cristalinas aumenta con el tiempo.
La mayoría de los estudios sugiere el análisis genético directo del gen ALDH3A2 ante la sospecha clínica [3,7,8], debido a su alta sensibilidad. Se han descrito más de 100 mutaciones diferentes causantes de síndrome de Sjögren-Larsson [9], sin que se haya observado una relación entre el genotipo y el fenotipo de la enfermedad. El 55% son mutaciones en homocigosis y el análisis mediante secuenciación del gen ALDH3A2 detecta el 95% de las mutaciones [3]. La gravedad del fenotipo en los pacientes con síndrome de Sjögren-Larsson es variable, incluso entre miembros de una misma familia portadores de idénticas mutaciones [3,8]. No existen muchos casos descritos, y la mayoría tiene poca o ninguna información clínica. En los últimos 30 años sólo se han notificado tres casos en España [4,7,10]. Nuestro caso clínico se suma a los pocos casos de origen español descritos en la bibliografía y aporta una nueva mutación, p.Glu331Lys, en el gen ALDH3A2. Aunque se trata de una variante que no se ha descrito previamente, varias evidencias indican que constituye una mutación patógena y no un polimorfismo: el análisis in silico mediante la herramienta PolyPhen [11] clasifica la variante como probablemente patógena. La mutación afecta a un aminoácido altamente conservado que forma parte del sitio catalítico de la enzima, y experimentos de mutagenia dirigida demuestran que mutaciones en este aminoácido anulan la actividad de la enzima [12].
Por tanto, y en primer lugar, ante un niño con parálisis cerebral espástica acompañada de lesiones en la piel debe considerarse el diagnóstico de síndrome de Sjögren-Larsson. En segundo lugar, debe realizarse un diagnóstico diferencial con otras leucodistrofias con lesiones cutáneas o enfermedades neuroictióticas, como la ictiosis ligada a X, la eritrodermia ictiosiforme congénita, el síndrome de Chanarin-Dorfman, la enfermedad de Gaucher de tipo 2 o la enfermedad de Refsum [3]. El diagnóstico definitivo del síndrome de Sjögren-Larsson lo proporciona el estudio del gen ALDH3A2, que además hace posible el consejo genético y el diagnóstico prenatal si fuera necesario.
Artículos relacionados: síndrome de sjögren-larsson, diagnóstico, investigación
Fuente: https://goo.gl/s2j9EP




